We’ve come a long way since the Human Genome Project. This effort to sequence the human genome for the first time was a tremendous achievement that lasted 13 (ish) years and cost $3 billion dollars. Thanks to this pioneering work and major advancements over the years it’s now possible to get your own genome sequenced for $300 in a few months.
That’s a 10,000x decrease in cost! If the price of homes decreased at a similar rate since 1990, the median home would cost $7.91 today.
In July (3 months ago) I ordered a whole genome sequencing kit from Nebula Genomics. I chose them because of the price, the good reviews, and the fact that they give you the ability to download all of your data when it is available.

I swabbed some cells off of my cheeks and sent the sample back in the pre-paid envelope, then waited. I read online that some users were waiting for ~9 months for their data to be available, but many were getting their results in 3 months.
WGS vs. SNP genotyping
SNPs (Single nucleotide polymorphisms) are differences in a single nucleotide when looking at a population. Humans have 99.9% of the same DNA as each other, so if you focus on what’s different you’ll see the most interesting information. Over time we have identified SNPs that are somewhat common in the population.
Companies like 23andMe and Ancestry offer SNP genotyping. Instead of sequencing your entire genome, they instead focus on the 690,000 nucleotides that vary the most from person to person. This is cost effective and can give you information about your heritage and some diseases that have been selected.
In contrast, whole genome sequencing (WGS) attempts to read your entire genome, all 6 billion nucleotides. This gives you all of the information that 23andMe does, plus a lot more. Mutations that are unique to you will be revealed using WGS.
The Results
At the end of September I received my results! I was able to login and download my data and view some results in Nebula’s online portal. There is roughly 100GB of data, so I was thankful for somewhat fast internet.
Nebula has a reporting library where you can see if you have the markers that have been identified in various genome wide association studies. For example, I am in the 96th percentile of nebula users to be predisposed to migraines, according to this study:

Nebula’s reports were fun to go through and included a few funny and rude results:
- ☕ 100% coffee consumption
- 😢 75% loneliness
- 😡 16% facial attractiveness
- 🏃♂️ 5% physical activity
But on the positive side, I am in the following percentiles for:
- 🦴 96% bone density
- 🧬 96% longevity
- 🦟 72% resistance to severe malaria
- 💤 0% insomnia
There were also a few interesting results that are good to be aware of and might cause me to make some lifestyle changes.
Gene analysis
Nebula gives you the ability to easily load variant data into gene.iobio. I obviously was interested in my CHM gene, so I checked that out:
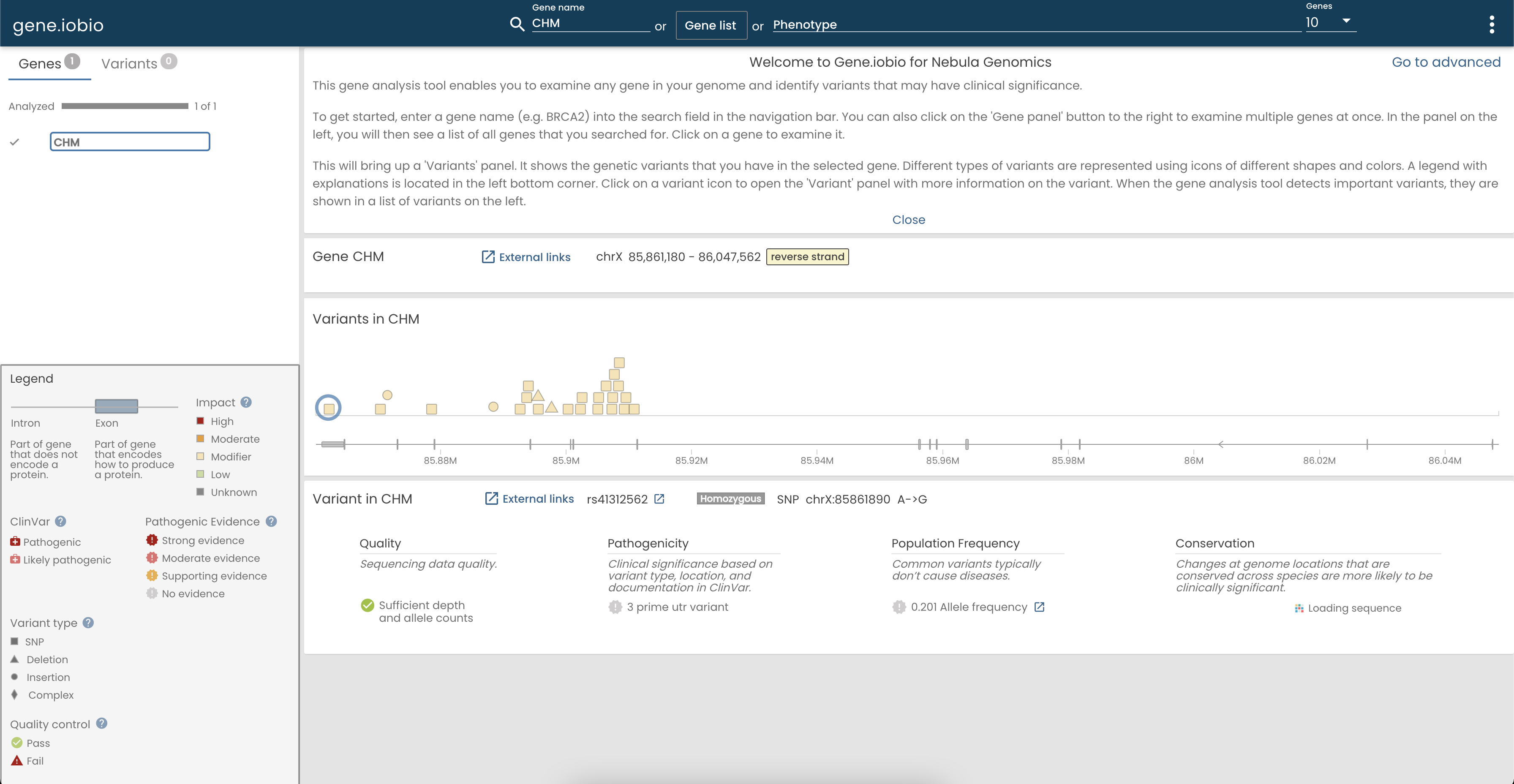
If you can see in that screenshot I have a bunch of variants at the beginning of my CHM gene, but after the variant at position 85,910,487, there aren’t any more variants. This is because my CHM causing mutation is a large deletion that spans exons 3 to 9. There are no variants in this section because I don’t have any DNA there!
Subscription
Part of Nebula’s business model is to require a subscription to access your data and reports online. The current, undiscounted prices for the subscription are
| Frequency | Price |
|---|---|
| Monthly | $19.99 |
| Yearly | $9.99/mo |
| Unlimited Lifetime Access | $200 |
I’m not sure how many new reports come in so I’m not sure if the subscription is worth it. There is a lot you can do with your data if you are so inclined that doesn’t require a subscription.
Using the data
I was able to load the VCF file into BigQuery using Google’s gcp-variant-transforms tool. This process worked smoothly for me and it has been interesting to query the data.
I have seen others recommend Promethease, so I will be checking that out next.
I am just getting started analyzing my sequencing data and I will post more about what I find along the way.
Note
None of this should be taken as medical or genetic advice. Talking to a genetic counselor is a good way to figure out if your variants are medically relevant or not.